

Transitive alignments (and why they matter)
by Ketil Malde; October 10, 2012
If you’re working with bioinformatics (or biology) at all, you can’t help but hear about “Next Generation Sequencing”. The evolution of sequencing technology is progressing rapidly, and the amount of sequences you get per dollar is accelerating faster than most growth curves, including disk space and computing power. This is great for biologists, who can easily (i.e. with a bit of standard lab work and a few thousand euros) sequence the genome of their favorite species, no matter how obscure. So the sequence data is piling up, and in a decade or so, storage for the results is going to be more expensive than the actual sequencing.
The problem
Unfortunately, there is another thing that doesn’t scale exponentially at all, and has been the bottleneck for some time: the analyst. When the Human genome sequence was finally published, it was the work of thousands over many years. Now, you are lucky if you can have a single post doc. dedicated to a genome project, and often you’ll just run a standard suite of analytical tools, and leave it at that.
This means two things:
- Automatic analysis becomes even more hugely important
- The pile of basically unidentified sequence data is growing faster and faster
The “analysis” in question often consists of (or at least starts with) identifying related sequences that we know something about. If two sequences are sufficiently similar, we believe they are derived from the same ancestor sequence, and that they are likely share some or all function. Clearly this is a gross approximation, but it is a starting point, and many, many sequences will never get any analysis beyond this point.
Now for this to make sense, we need some target sequences that we actually do know something about. There aren’t that many, one example is SwissProt, a database of sequences with curated annotations. The downside is that it only contains about half a million sequences, about two percent of the total known proteins. And very few species are anywhere near complete - unless you are interested in something very close to human, mouse, fruit fly, or yeast, you are basically out of luck.
Dealing with it
The obvious answer is to go indirectly. Take the set of all proteins (from UniProt or NR), align each sequence against SwissProt, and annotate it with its best match. Then compare your sequences against this large, indirectly annotated database. This is a fairly typical approach, and for instance Gene Ontology does this on a large scale, and out of the millions of assigned terms describing function, cellular localization, and processes, only a few thousand are actually based on experimental data - the rest are IEA - inferred by electronic annotation. The good thing is that they tell you about it, with many other databases, annotation is assigned by the submitter on whatever basis she feels appropriate, a basis which is then quickly forgotten.
There is another alternative that has become quite popular: Take all sequences that we know are similar, and build a stochastic model from them, and search against that. The canonical tool here is HMMER, which uses hidden Markov models to model sequence sets. It is typically used with the Pfam set of protein motif models. This is more sensitive than searching directly, but requires more curated data to build the HMMs, and at least one paper found that for small sets of sequences, just using the best BLAST score was superior.
Transitive alignments provide a third way. It can be seen as sort of a middle road - it uses the wealth of uncurated data to build better pairwise alignments, but it does this directly, there is no need for explicit (or implicit, for that matter) models - Markov or otherwise. One advantage to this is that unlike HMMs, simply adding to the pool of uncurated sequences will improve transalign searches for free, and this is of course inevitably happening.
A sketch of the algorithm
The program works by first doing alignments against an intermediate, uncurated database. Remember that an alignments is basically just a set of links between positions in the target (in this case intermediate) and query sequences. Now, we already have alignments from the intermediate database to the target database (SwissProt, say). So for each position in an intermediate sequence, we can identify a corresponding position in a target sequence. What remains is to make this transitive: so if query position p is aligned to intermediate position q, which is mapped to target position r, we make a transitive alignment mapping p to r.
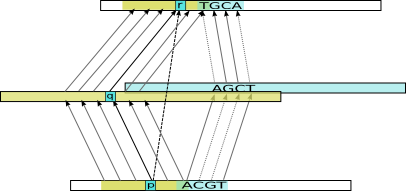
Example transitive alignment constructed from combining the indirect alignments for two intermediate sequences (yellow and blue)
Doing this for many intermediate sequences, we get a lot of possibly inconsistent mappings, so this is cleaned up in a method very similar to standard sequence alignment, and the set of position pairs that maximize the total score is produced as our final transitive alignment. The final alignment will incorporate the best evidence via many intermediate sequences, and this often makes it possible to extend the alignments through regions where the target and query sequences have diverged substantially.
Results
So that’s all fine, but does it work? I hope to get a paper out Real Soon Now, but I’ll include one figure here. This is using data from the SCOP database. SCOP takes proteins with known 3D structure, and classifies them hierarchically - most similar are proteins in the same family, families are grouped into superfamilies, and superfamilies into folds, which finally are collected in classes.
Protein 3D-structure is regarded as better conserved than sequence, and sequence alignments are considered to be ineffective beyond a twilight zone around 30% identity.
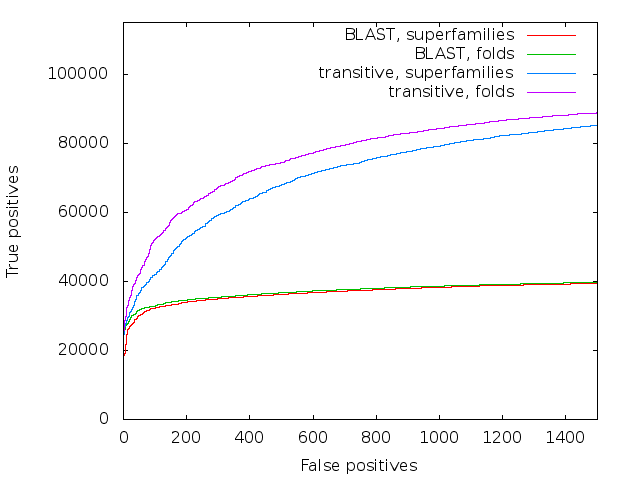
Accuracy of direct BLAST and transitive alignment BLAST on the SCOP database
We took SCOP40 (a subset of SCOP where the sequences are at most 40% similar to each other), and simply aligned all-against-all, sorting the results on score. The plot is a standard ROC curve, it shows the false positives (that is, a match between two sequences that are in different superfamilies/folds) along the x-axis, and the true positives (matches between sequences in the same superfamily/fold) on the y-axis. It is remarkable that not only does transalign perform vastly better on superfamilies - as long as you go past the very similar matches at the start, it has about twice the sensitivity of direct blast - but it also manages to find many fold relationships between superfamilies, where direct blast finds practically none.
Why does it work?
Most statistics analysis of sequence alignments and searching assumes that sequence databases are random - of course, they are no such thing. Sequences have patterns: some things happen to work out biologically, other things don’t. Also, many sequences are closely related, derived from a not-to-distant ancestor. So for any relationship between two sequences, the huge databases of raw sequence provides a lot of context for that relationship. One way to look at it is that we use intermediate sequences as sort of a bridge - but transitive alignments go beyond that by piecing together the different alignments. This way, we can use different intermediate sequences for different parts of the alignments. For instance, the closest intermediate overall may has suffered some heavy mutation in one part of the sequence, and then more distant relatives may produce a better alignment in that region.
In any case, the method is really very simple, and it performs much better than anything else I’m aware of. What’s not to like?
comments powered by Disqus