

Thoughts on phylogenetic trees
by Ketil Malde; February 15, 2015
An important aspect of studying evolution is the construction of phylogenetic trees, graphically representing the relationship between current and historic species. These trees are usually calculated based on similarities and differences between genetic material of current species, and one particular challenge is that the topology of the resulting trees depend on the selection of genes used to construct them. Quite often, the species tree based on one set of genes differ substantially from the tree based on another set of genes.
The phylogenetic tree is usually presented as a simple tree of species. The end points of brances at the bottom of the tree (leaves) represent current species, and branching points higher up (internal nodes) represent the most recent common ancestor, or MRCA, for the species below it.
A very simple example could look something like this:
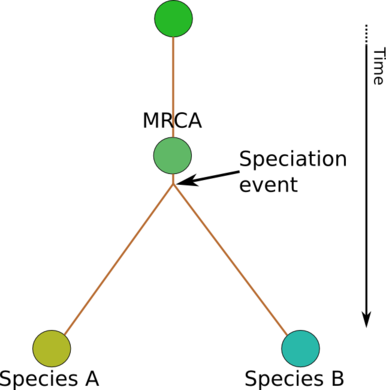
Evolution as a tree of species
Here you have two current species, and you can trace back their lineage to a MRCA, and further back to some ancient origin. Varying colors indicate that gradual change along the branches has introduced differences, and that the current species now have diverged from each other, and their origin.
This representation has the advantage of being nice and simple, and the disadvantage of being misleading. For instance, one might get the impression that a species is a well-defined concept, and ask questions like: when the first member of a species diverged from its ancestor species, how did it find a mate?
Species are populations
But we are talking about species here - that is, not individuals but populations of individuals. So a more accurate representation might look like this:
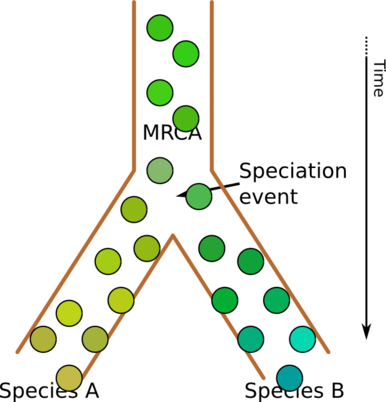
Evolution as a tree of populations
Circles now represent individuals, and it should perhaps be clearer that there is no such thing as the “first” of anything. At the separation point, there is no difference between the two populations, and it is only after a long period of separation that differences can arise. (Of course, if there are selective pressure favoring specific properties - perhaps redness is very disadvantageous for species B, for instance - this change will be much quicker. Look at how quickly we have gotten very different breeds of dogs by keeping populations artificially separate, and selecting for specific properties.)
The so-called “speciation” is nothing more than a population being split into two separate parts. Typically, this can be geographically - a few animals being carried to Madagascar on pieces of driftwood - but anything that prevents members of one branch from mating with members of the other one will suffice. At he outset, the two branches are just indistinguishable subpopulations of the same species, but if the process goes on long enough, differences between the two populations can become large enough that they can no longer interbreed, and we can consider them different species.
In practice, such a separation is often not complete, some individuals can stray between the groups. In that case, speciation is less likely to happen, since the property of being unable to breed with the other group represents a reproductive disadvantage, and it would therefore be selected against. In other words, if your neighbor is able to have children with more members of the population than you, his chances of having children are better than yours. Your genes get the short end of the stick. Kind of obvious, no?
Populations consist of genes
But we can also view evolution as working on populations, not of individuals, but of individual genes. This is illustrated in the next picture:
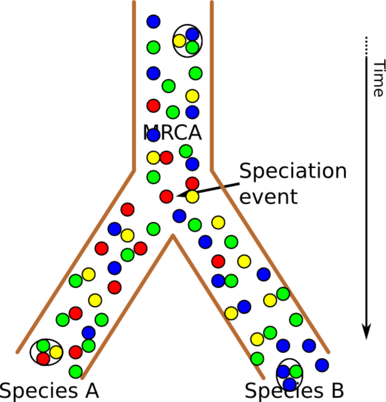
Evolution as a tree of gene populations
The colored circles now represent genes, and an individual is here just a sample from the population of genes - illustrated by circling three gene circles. (Note that by “gene”, we here mean an abstract unit of inheritance. In other fields of biology, the word might be synonymous with a genetic region that codes for a protein, or is transcribed to (possibly non-coding) RNA.)
Here, we see that although the genes themselves do not change (in reality they are subject to mutations), the availability of the different genes vary over time, and some might disappear from one of the branches entirely - like red genes from species B here. This kind of genetic drift can still cause distinct changes in individuals.
Ancestry of individual genes
Each individual typically gets half its genes from each parent, one fourth from each grandparent, and so on, so after a few generations, all genes come from essentially different ancestors. This means you can calculate the MRCA for each gene individually, and this is exactly what has been done to estimate the age our “mitochondrial Eve” and “Y-chromosomal Adam”. Here is the example lineage for the green gene:
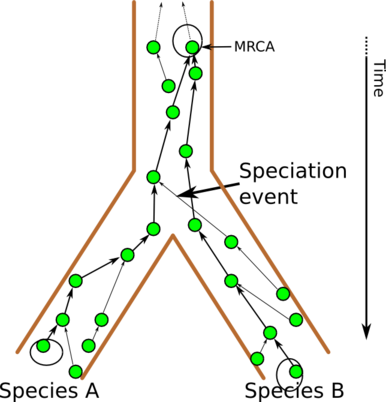
Lineage and MRCA for the green gene
We see that the green-gene MRCA is much older than the speciation event. In addition, each gene has its unique history. This means that when we try to compute the MRCA, different genes will give different answers, and it can be difficult to construct a sensible consensus.
For example, bits of our genome appear to come from the Neanderthal, and those bits will have a MRCA that predates the time point where Neanderthal branched from H. sapiens (possibly 1-2 million years ago). (Interestingly, “Adam” and “Eve” are both estimated to have lived in the neighborhood of 200000 years ago. This means that although 20% of the Neanderthal genome is claimed to have survived, all Neanderthal mitochondria and Y-chromosomes have been eradicated.)
comments powered by Disqus